

One-photon microscopy has been a crucial tool for advancing our knowledge in cellular biology; but with all techniques in science, there are restrictions. Two-photon microscopy aims to ameliorate many of these limitations by expanding fundamental principles in conventional imaging techniques. In this article we will discuss the differences between one and multiphoton microscopy and the advantages that come with each of these methods.
Contents:
• General overview of fluorescence microscopy
• How does one-photon microscopy work?
• How does two-photon microscopy work?
• Advantages of two-photon microscopy
• Advancements in multiphoton microscopy
General overview of fluorescence microscopy
Conventional fluorescence microscopy works by evenly illuminating a specimen from a given light source. The light that passes through the sample can be modulated by filtering out a particular wavelength of light to visualize multiple colored fluorophores across channels in a single field of view. Once the sample is excited with this wavelength, the emitted light is collected by the camera of the microscope, generating an image of your specimen. While this method has been indispensable across multiple fields of science, it is limited by the resolution and contrast quality that can be achieved. One main reason for this is that since the entirety of the sample is illuminated, fluorescence is collected from regions that are not within the focal plane of interest causing high background intensity.
Confocal microscopy is one of the most popular imaging techniques used in modern scientific and industrial laboratories to try and minimize the amount of background fluorescence that is collect from out-of-focus regions. The basic concept of this method is to increase the optical resolution and contrast by illuminating and blocking the collection of out of focus light emitted from the sample. This principle was a major advancement in imaging that mitigates many of the caveats of wide field fluorescence microscopy. Confocal microscopy reduces the collection of unwanted fluorescence through the use of single point illumination in combination with a pinhole to filter out signals that are unfocused. Together these properties permit the collection of light only from sections of the specimen extremely close to the focal plane (Figure 1a). This allows for optical sectioning of the sample and three-dimensional reconstruction by stacking images collected in in a z-projection.
How one-photon microscopy works
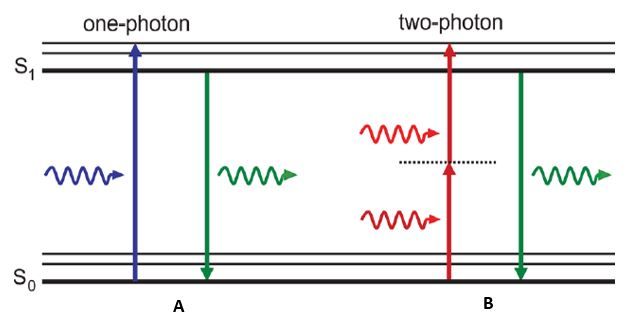
In these traditional microscopy methods, fluorescence is produced by shining a light of a corresponding wavelength to your fluorophore. This wavelength must be the appropriate amount of energy to excite electrons in your fluorophore to a higher energy level state. Once these electrons are elevated, they become unstable and can only stay at this excited state for a short period of time. Electrons then return back to their lower energy level but during this event energy is released in the form of a photon. Since energy is lost during this return to ground state, the light produced is at a higher wavelength and subsequent lower energy than the light that was used to excite your fluorophore. This process uses a single photon of light to cause this excitation and fluorescence in traditional fluorescence and confocal microscopy methods (Figure 2a).
How two-photon microscopy works
The main difference between a one-photon microscope and a two-photon microscope is exactly what it sounds like: one-photon uses one photon and two-photon uses two photons. This concept suggests that two photons can be used to excite a single molecule from ground state to a higher energy state in a single quantum event. In this case the energy difference required to cause electrons to jump energy states is equal to the sum of the energy absorbed between the two photons. Since the correlation between energy and wavelength is linear, each photon of light has double the wavelength when compared to the light that is used excite the same fluorophore in single-photon methods (Figure 2b). Even though two photons are being used to excite electrons to a higher energy state, still only one photon is emitted when the electron returns to ground state, resulting in an equivalent wavelength as the single-photon fluorescence method.
Advantages of two photon microscopy
Since two-photon microscopy requires that two photons of light excite a fluorescent molecule to its higher energy state, fluoresence is only produced when these photons interact with this molecule within 1 femtosecond of each other. This means that light is only collected from a single point closest to the focal plane and the above and below planes are not illuminated (Figure 1b). For this reason a pinhole is not required in two-photon microscopy where it is required to filter out unfocused light during confocal microscopy. The following advantages are the result of this mechanism and why two photon-microscopy may be better suited for certain biological imaging assays.
Photobleaching:
Both confocal microscopy and two-photon microscopy have optical sectioning capabilities to visualize three-dimensional reconstructions of your specimen. However, this is achieved through different means, either through use of a pinhole (confocal) or point illumination (two-photon) as previously discussed. In confocal microscopy, excitation light continues generate fluorescence throughout the entire specimen. In this case, light is absorbed by the tissue which can cause photobleaching of your signal and also photodamage to the tissue you are visualizing throughout the entirety of your sample. This can cause complete quenching of your fluorescent signal and even kill the tissue you are visualizing if excited for long enough.
In the case of two-photon microscopy, the fluorescence that is generated is much more localized than in conventional fluorescence microscopy. Fluorescent objections that are not within the focal plane are not excited since the only objects that are being excited are at the position of where the two-photon excitation is focused. This is where in contrast a confocal uses a pinhole in order to filter light and achieve optical sectioning. Since in this method, fluorescence is restricted to only to the focal plane. This drastically localizes the degree of photobleaching to a much smaller region compared to confocal microscopy .
Imaging Depth:
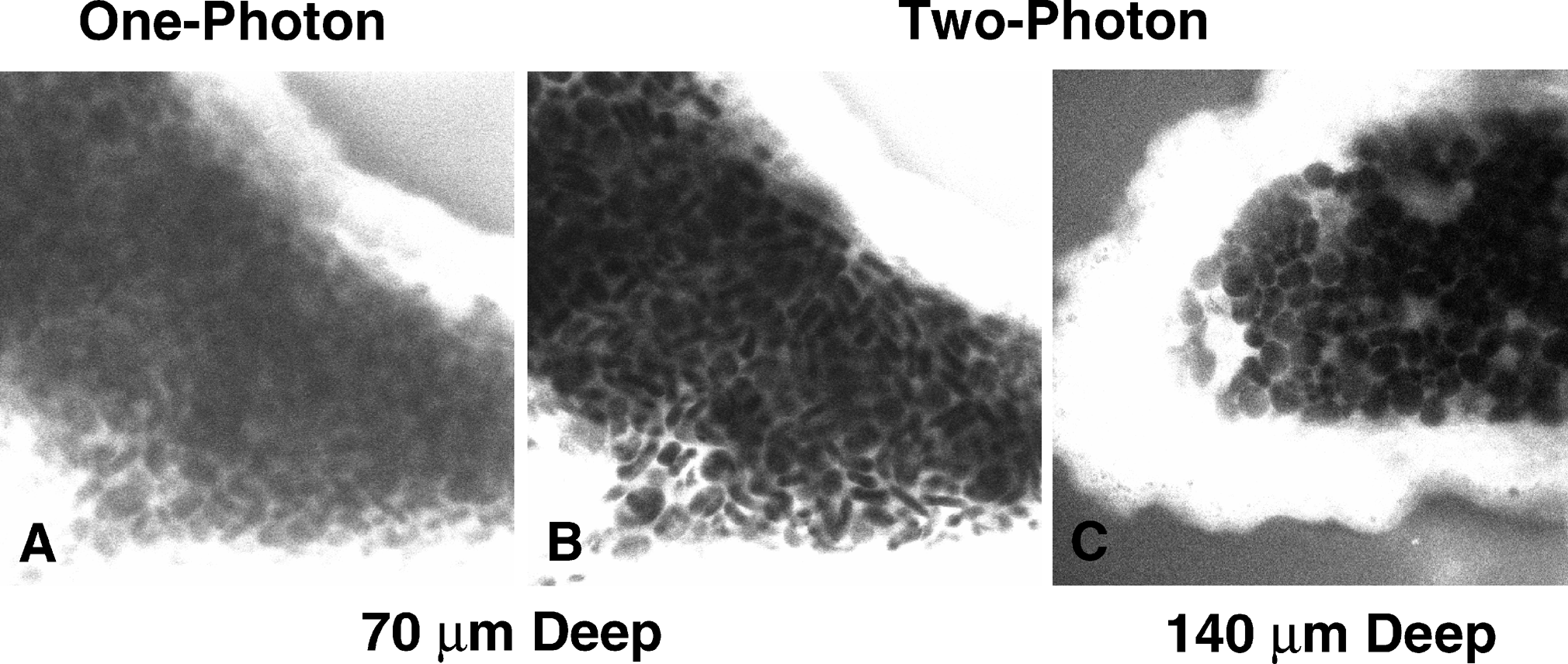
One of the main advantages two-photon microscopy has over conventional methods is that you can greatly increase the depth of imaging within thick tissue samples. The main property that controls how deep one can image is light scattering. When light passes through a sample to generate fluorescence in a specimen, light will scatter in a heterogenous manner. The imaging depth of a sample is directly proportional to the amount of light that will scatter because there is increased material for the light to have to pass through. Again, the pinhole on a confocal filters out much scattered light to only collect fluorescence at that imaging depth. However, as the imaging depth increases there is a greater likelihood that this light will also scatter outside of the confocal pinhole and will reduce the signal at that plane. Another caveat is that since fluorescence is generated throughout the specimen and more light is scattered from above and below the focal plane, this increases the likelihood of light scattering through the pinhole from out of focus regions. The combination of these events causes decreased signal of your image and increased background at greater imaging depths with confocal microscopy.
Two-photon microscopy has multiple properties that ameliorates many of these effects to achieve greater imaging depths. Since the way fluorescence is generates is by focusing two-photons of light at a single position to cause excitation and generate fluorescence, the same property applies for light that can be collected as background signal. In order for background fluorescence to be produced, two excitation photons must scatter at the exact same position. Since the odds of this event occurring at great depths in heterogenous biological samples, there is no background signal and drastically boosts the contrast of your image. Another property that increases imaging depth has to do with the wavelength of light that is used to excite your fluorophore. As previously described, two-photon microscopy uses lower energy and subsequently greater wavelengths of light compared to confocal microscopy. This wavelength used is infrared light which scatters much less when compared to the visible wavelengths used in conventional methods. This allows a greater proportion of light to reach larger imaging depths allowing for visualization of regions within thick samples (Figure 3).
Optical stimulation assays:
For all of the advantages above regarding imaging depth, excitation localization, and reduced phototoxicity, two-photon microscopes can be used for novel assays that involve photo activation within regions of tissue. Photochemical reactions can be carried out in the context of activation of caged molecules. Caged molecules are well established tools which enclose a molecule of interest which can then be locally released via light activation. Some of the most common uses of this technique appear when studying the effects of neurotransmitters in populations of neurons. For example, one can record the electrical activity from a neuron deep within the brain or a mouse and simultaneously pulse light to uncage the neurotransmitter glutamate and study how the recorded neuron changes its activity in a temporal manner.
Another useful technique that can be used with a two-photon microscope is the control of electrical activity of cells via photo-activation of ion channels. This field is also a major advancements in studying neuronal circuitry across brain regions. This works by expressing an optically activated ion channel such as ChannelRhodopsin in a subset of neurons. You can then record the activity at the presynaptic and post synaptic populations and photoactivate the ChannelRhodopsin expressing cells and study the effect their activation has on this particular circuit. This allows for much finer control over which cells to activate when comparing to glutamate uncaging experiments. Though both of these photo activation techniques are possible with confocal microscopy, two-photon microscopy adds another degree of spatial precision and stimulation depth when it comes to assaying live specimens.
Further advancements in three photon microscopy
The jump from one two two photon on microscopy has enabled great leaps in biological sciences for imaging in thick and live specimens. Adding one more photon can potentially be even more advantageous. In three-photon microscopy, the same concepts apply as in two-photon microscopy; in this case, three photons must simultaneously excite a molecule to generate fluorescence. Consequentially, the wavelengths of light used must be three times longer than one-photon versus twice as long in two-photon. These longer wavelengths allow for even deeper penetration of light to excite fluorophores at greater depths in intact tissue.
A main difference with this approach is that it requires a specialized high power laser to generate higher photon density and longer wavelengths. One approach is using a optical parametric amplifier which splits one wavelength of light into two. One of these wavelengths will be shorter and can be applied for two photon approaches while the longer one can be used for three photon imaging. Advancements in this field can have major implications in non-invasive, live cellular imaging within a living organism.