
RNA fluorescent in-situ hybridization (RNA-FISH) has a notorious reputation among biologists—many of us have been warned off this technique by horror stories of difficult protocols and low success rates. However, FISH remains the gold standard for visualizing RNA expression and localization in cells, tissue sections, and whole organs. Major methodological advances in ease of use, sensitivity, and reproducibility have made it easier than ever to incorporate RNA-FISH into your next paper. In this article, we’ll do a deep dive into:
- How RNA-FISH works
- Basic steps in RNA-FISH protocols
- New developments in RNA-FISH
- RNA-FISH in spatial sequencing
In-situ hybridization (ISH), which underlies all FISH approaches, takes advantage of the thermodynamic properties of nucleic acids. Complimentary single-stranded oligonucleotides will anneal to each other when conditions favor binding and form double-stranded RNA, DNA, or RNA:DNA hybrids. Therefore, if you want to find your favorite mRNA (or DNA) sequence within a cell, you might try synthesizing a complementary oligo sequence, tagging it with some marker, and applying it to your fixed cells. Hopefully, the tagged oligo will bind your RNA of interest and allow you to visualize it using a signal amplification method. This was the approach piloted by Gall & Pardue in 1969 to visualize Xenopus oocyte DNA, using radioactive ISH probes. Later innovators swapped out radioactive probes (expensive and hazardous) for fluorophore-tagged probes, coining FISH. The use of FISH for RNA detection dates from 1982, and increasing technical advances led to the development of single-molecule FISH (smFISH), where individual mRNA molecules are visualized. Many variations on the basic FISH protocol have been described and may be optimized for different niche applications. We’ll start by describing the common elements of an RNA-FISH experiment: sample preparation, probe hybridization, and washing. We'll also discuss considerations for probe design below.
Basic steps in RNA-FISH
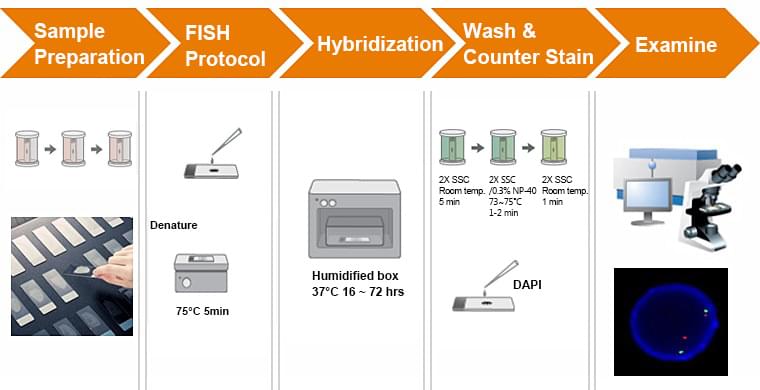
Sample Preparation
Tissue fixation and permeabilization are the two most important aspects of sample preparation for FISH, significantly impacting the quality of the data extracted. Fixation is commonly achieved using 4% paraformaldehyde (PFA), which should be prepared fresh for each usage as PFA in solution forms polymers with long-term storage. An alternative is to use a 1:10 dilution of formalin, which does not need to be prepared fresh each time. The incubation period for fixation is highly variable and dependent on the specific tissue—optimized protocols range from 6h for whole mouse brains to 10min for bacterial cells. This is really where trial-and-error or a literature search are necessary. Similarly, fixation temperature affects the quality of the final signal and requires optimization. Although formaldehyde-based fixation is most common, ethanol-based fixation is also used. This involves the use of ethanol or methanol to dehydrate cells and tissues, and can also be used in combination with formaldehyde.
Permeabilization is usually achieved post-fixation using a detergent such as Tween-20, SDS, or Triton X-100. Concentrations of detergents range from 0.1% to 4% (the bulkier the sample the more detergent used—think whole brain vs sectioned tissue). For applications other than eukaryotic cells a proteinase treatment is often applied as well, especially when the penetration of oligo probes is thought to be an issue. Proteinase K treatment nonspecifically digests RNA-binding proteins, potentially making the target sequences more accessible, but can also destroy the integrity of the tissue when over-incubated.
Probe Hybridization
In this step, RNA-specific probes are applied onto the fixed and permeabilized sample. Since hybridization probes are applied onto the sample in solution and rely on diffusion kinetics to enter the cell, incubation times are relatively long– on the order of hours to days. Additionally, longer molecules move slower, so the longer the probe, the longer the minimum incubation. Specialized hybridization buffers are used to increase the chances of successful probe binding. Most buffers include the following components:
· Formamide: reduces the energy barrier for nucleic acid binding allowing hybridization to occur at lower temperatures
· Vanadyl-ribonucleoside complex: an RNase inhibitor which protects RNA-based probes as well as the target RNA
· Dextran sulfate: a crowding agent that encourages proximity of probes and targets
· Bovine serum albumin (BSA): blocks nonspecific probe binding
· Sheared salmon sperm DNA or E. coli/yeast tRNA: also blocks nonspecific probe binding
The other major variables in the hybridization reaction are salt concentration and hybridization temperature. If you’ve ever done a PCR reaction, you’ll recognize these factors—the same concepts of nucleic acid interaction apply. All of these variables must be optimized for a given probe, although basic starting points are as follows: keep the salt concentration constant (750 mM NaCl, 87.5 mM sodium citrate) and maintain a pH between 7.0 and 8.5, while trying different hybridization temperatures to find the optimal signal with least background/nonspecific binding in a time frame of 12-24hrs. In general, the longer the probe the higher the required hybridization temp.
Washing
Following hybridization, several wash steps are necessary to remove nonspecifically bound probe and remove background signal. In general, washes move from higher to lower salt concentration and lower to higher temperature to force removal of weakly bound probes that cannot withstand the stringency of the washing steps. Additional sample processing can be done at this step as well to improve the quality of the final staining, including quenching autofluorescence with a solution of Sudan Black dye and clearing tissue using organic solvent or commercial reagents use as ClearT. Nuclear staining is also often performed as well prior to FISH signal detection. This is typically done on an epifluorescent or confocal microscope at high magnification.
Probe Design Variables
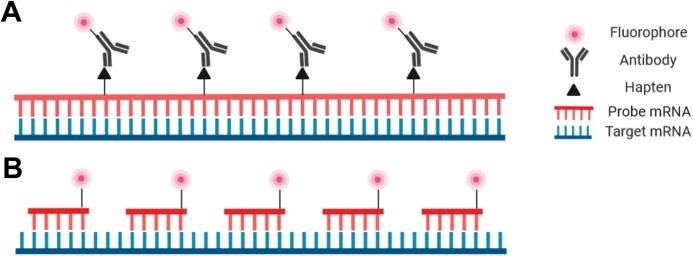
Probes for RNA-FISH are DNA, RNA, or cDNA strands that range in length from 20 bp to over 1000 bp. The most important criterion is sequence specificity to the target RNA. A typical probe for generalized RNA-FISH is a relatively long (500-1000bp) ssRNA riboprobe produced via in vitro transcription. Riboprobes are usually detected using a fluorophore-conjugated antibody, but DNA probes can also be directly conjugated to a fluorophore. An additional increasingly used probe option is the oligonucleotide probe—actually a set of small DNA oligos that each bind to a specific region of the target RNA. This type of probe offers the highest specificity and is particularly useful for tissue types in which larger probes have difficulty penetrating, as well as for applications that are highly specificity-dependent such as identifying splice variants and single nucleotide polymorphisms (SNPs). Several bioinformatics tools have recently been developed to simplify the process of designing oligo probes, among them ProbeDealer and OligoMiner. Intelligent design of hybridization probes to avoid repetitive elements, nonspecific binding, and cross-hybridization is critical for successful staining.
Controls and Troubleshooting
The last step in the FISH protocol is imaging the final stained sample to (ideally) obtain an image of the target RNA staining pattern. Not as simple as it sounds though– a commonly encountered issue during FISH protocol optimization is uncertainty around whether a positive signal reflects actual RNA distribution (rather than nonspecific probe binding) or whether lack of signal represents true absence of target RNA in the sample or inappropriate staining protocol. Controls are therefore a very important aspect of a well-designed FISH experiment. As in qPCR, a well-expressed constitutive “housekeeping” RNA such as actin can be used as a positive control in order to rule out reagent or method issues. Conversely, a negative control can be generated using reverse complement probes (“sense” probes) which should not be able to bind to the target RNA like normal complementary “antisense” probes. Treatment of the sample with RNase and/or DNase can also rule out nonspecific binding of fluorophore-tagged antibodies since probes should not be able to bind these samples. For more technical detail on controls, troubleshooting, and all the steps described above, see this excellent review!
New developments in RNA-FISH
The evolution in FISH technology has come on multiple fronts. In the quest to identify target RNA with greater and greater resolution, advances in probe design and signal amplification have made it possible to detect single RNA molecules in fixed cells. Given that many important mRNAs are produced at very low copy number (e.g. transcription factors), achieving high resolution is critical for accurate quantification. This breakthrough was first reported in 1998, and is now called single-molecule FISH or smFISH. smFISH takes advantage of the multiple oligonucleotide probe approach described above for the highest signal-to-noise ratio possible. Variations on smFISH have been developed in the past 20 years—everything from smiFISH to FISH-STICs—with the main improvements occurring at the signal readout stage through development of novel probe and amplification designs. This review covers the relevant protocols that have been developed in the smFISH space.
The other main thrust of method development in RNA-FISH centers around multiplexing, or imaging multiple target RNAs on the same sample. Multiplexed FISH can be achieved with sequential rounds of rounds of imaging, label removal and relabeling as in the technique DNA-Exchange. Other approaches, such as MERFISH and seqFISH, use multiple oligonucleotide probes for each transcript imaged and multiple rounds of hybridization and stripping to generate unique fluorescent barcodes for each gene detected. The fluorescent pattern of the sample is then deconvoluted by software, generating gene expression patterns for up to 10,000 RNA targets.
Improvements in RNA-FISH staining resolution and multiplexing have contributed to the development of whole new method field: spatial sequencing. We discuss the theory and practice of spatial sequencing below.
RNA-FISH in spatial sequencing
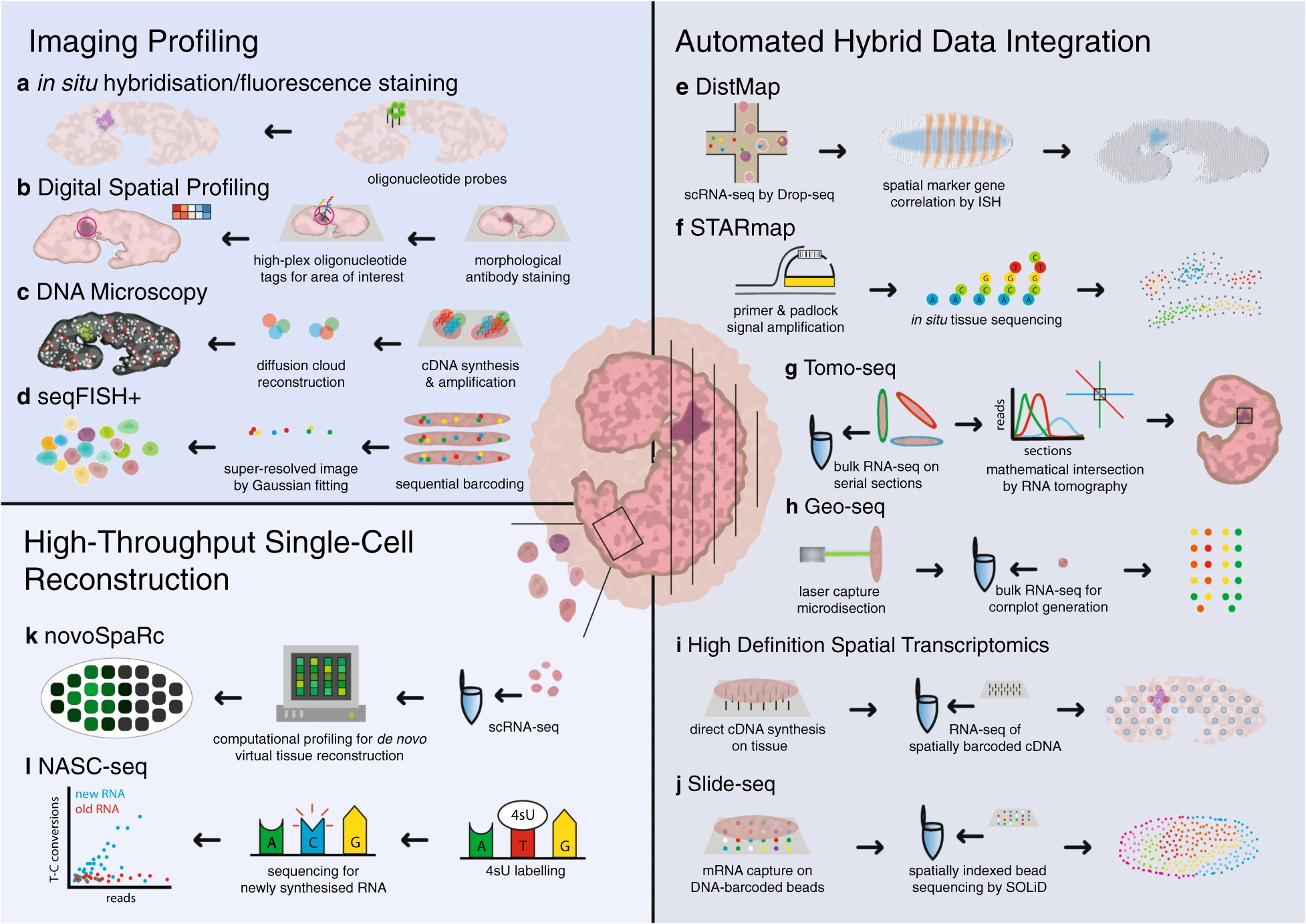
Spatial sequencing, also known as in situ transcriptomics, is the younger sibling of single-cell RNAseq (and 2021 method of the year!). Spatial sequencing allows researchers to map transcripts onto cellular and tissue architecture and understand the context of mRNA expression. The more RNA targets we can image at once the closer we get to capturing the entire transcriptome expressed in a given sample. Similarly, as resolution of single RNA targets increases, the closer we are to resolving the transcriptome of single cells in situ. Although fully spatially resolved single-cell transcriptomes are not yet possible, many researchers in this fast-moving field are optimistic that it’s coming soon. Several spatial sequencing methods are based on RNA-FISH—MERFISH and seqFISH, discussed above, fall into this category, as are other methods such as RNAscope. Other approaches to spatial sequencing include in situ sequencing (for example, see FISSEQ) and in situ capture using microbeads tagged with mRNA-capturing primers (see: Slideseq).
A continuous stream of new spatial sequencing techniques and refinements of existing methods appear every year, making it difficult for a newcomer to the field to determine which approach is best for a given application. The SpatialTx project, spearheaded by researchers at the Chan-Zuckerberg Institute (CZI) in San Francisco, aims to provide some clarity by benchmarking 19 different spatial transcriptomics methods on adult and developing human and mouse neocortex samples. SpatialTx is ongoing, so stay tuned for the findings! In a parallel effort, CZI scientists have developed an open-source computational pipeline for spatial transcriptomic analysis called Starfish, meant to simplify and standardize analysis across different FISH methods.
Commercially available kits for spatial transcriptomics are now widely available from several different sources, all based on different approaches to target RNA capture. Here’s a summary of several popular options:
· GeoMx Digital Spatial Profiler (Nanostring): This technology uses UV-cleavable hybridization probes and downstream sequencing to identify gene expression in a given region of interest in a tissue sample.
· RNAscope (ACD): This RNA FISH-based method uses a proprietary Z-probe design to improve signal-to-noise. Also available is miRNAscope, used for visualizing small RNAs.
· Visium (10x Genomics): Samples are placed onto glass slides covered in mRNA capture probes labelled with spatial barcodes, and bound target RNA is reverse-transcribed into cDNA for downstream sequencing.
With all the techniques, modifications, and commercial solutions out there, any researcher looking to use RNA FISH has lots of options and a good chance of success. Happy FISHing!